小儿多囊肾原因何在?

(一)发病原因多囊肾的病因是基因缺失。其中成年型多囊肾常是由于16号染色体的基因缺失,偶然是由于4号染色体的基因缺失,是外显率为100%的显性遗传,因此单亲的染色体缺失将使其子女有50%的可能性遗传该疾病。婴儿型多囊肾是常染色体隐性遗传,父母双方均有该病的基因改变才能使其子女发病,发病概率为25%。
(二)发病机制
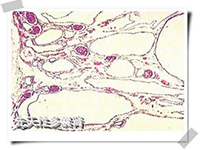
1.发病机制 肾囊肿衍生于肾脏的上皮结构,原发于肾小管和包氏囊。所有的肾囊肿都有某些常见的结构成分,包括上皮质、膨胀的囊肿内含肾小球的滤液。
(1)常染色体显性遗传型PK:95%以上的典型患者是由第16对染色体短臂上的异常基因引起,加上感染和中毒作用于小管,激发囊肿基因改变了小管细胞代谢,直接造成上皮细胞坏死,造成梗阻亦促进细胞增生致囊肿形成。
(2)常染色体隐性遗传型PK:是由DNA突变引起,但缺陷的等位基因的染色体位点尚不清楚。患儿之父母并不患病,但都携带这种病的基因才能使其子女发病,此病罕见。
2.病理变化
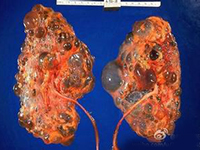
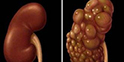
(1)婴儿型多囊肾(infantile polycystic kidney):是常染色体隐性遗传,均伴肝脏病变,虽然主要见于年幼儿,但也可发生于年龄较大的儿童及成人。 双肾显著增大,外形保持正常,表面光滑,但胎儿肾脏的分叶状态较正常肾更明显。因肾的皮髓质被小囊肿所侵犯,故切面呈海绵状或蜂窝样。组织学检查,肾实质被多数与肾表面成直角排列的长的囊肿所代替。在被膜下可看到少数正常的肾小球及曲管。肾功能检查显示囊肿是肾单位的功能部分,显微解剖可见囊肿为扩张的集合管。输尿管及膀胱发育正常。严重病例,由于胎儿尿少导致膀胱发育不良。根据症状出现时年龄,婴儿型多囊肾可分为4个临床亚型: ①围产期(perinatal period)型:由于婴儿腹部膨隆,产程往往长而不顺利,可见小儿循环不良及呼吸困难,呈现典型的Potter面容。有时因并发肺发育不全,纵隔积气而出现发绀,显著呼吸困难,可于出生时或生后不久死亡。有些婴儿出生时可触及巨大肾脏,生后有尿毒症、脓尿、血尿及高血压,围产期可能存活。这些婴儿肾脏的90%或更多的肾组织为囊性或发育异常。静脉泌尿系造影双肾不显影,肾超声波检查可证实大而多囊的肾。排尿性膀胱尿道造影显示膀胱正常,无反流,无尿道梗阻。如出生时就有尿毒症则预后不良,死产的百分率高,绝大多数尿毒症婴儿均于围产期或生后3个月内死亡。 ②新生儿型:在新生儿期存活者,常有进行性尿毒症、高血压,并可触及双侧肾增大。经肾超声波检查,囊肿病变范围广泛,约60%肾单位发育异常,多于生后6月内死于尿毒症。有些患儿经适当限制饮食中的蛋白质,治疗肾性酸中毒、高磷血症及高血压可使尿毒症减轻,肾功能改善,病儿可存活到儿童期,但肝脏的病变随年龄增长逐渐显现。③婴儿型:有25%~50%肾单位有囊性肾发育异常,临床表现为生长迟滞、进行性尿毒症以及儿童期的肝功能衰竭,在出生时难与新生儿型区别,只是婴儿型在婴儿期没有进行性尿毒症。 ④童年型:由于双侧肾囊性病变小于10%,而肝脏病变广泛,故在10~20岁时表现肝病变,只有做尸检时才偶然发现有多囊肾。
(2)成人型多囊肾(adult polycystic kidney):本症是常染色体显性遗传,约3%病例于小儿期就开始有症状,但在小儿引起肾功能衰竭者罕见。双侧肾增大,大小不规则的囊肿散在于皮质及髓质,夹杂有正常肾实质,囊肿可在肾单位或集合管的任何部位。肾小球囊肿是成人型多囊肾在早期的一个特点。成人的并发症(出血、结石及感染)不常见于小儿,也往往不严重。散在局灶性肝囊肿只占成人患者的1/3,不产生功能障碍,脾及胰囊肿不常见。
- 上一篇:导致肾囊肿的因素有什么
- 下一篇:胎儿单侧多囊肾的原因是什么呢
- 本文延伸阅读
- 2016-04-25
- 2016-04-25
- 2016-04-25
- 2016-04-25
- 2016-04-25
相关文章
- 多囊肾产生的原因
- 小儿多囊肾的病因有哪些
- 多囊肾是怎么引起的
- 多囊肾是怎么回事?
- 多囊肾有哪些常见的病因?
- 多囊肾的病因究竟是什么呢?
- 多囊肾是怎么引起的?
- 多囊肾病因包括哪些因素
- 诱发肾囊肿的因素有哪些
- 引起多囊肾的病因有哪些
- 多囊肾疾病的病因你了解吗
- 多囊肾的发病原因有哪些


- 多囊肾分期有什么规律?
- 目前尚无任何方法可以阻止疾病的发展,早期发现,防止并发症的发生与发展,及时正确地治疗已出现的并发症至关重要,下面小编就来详细的介绍一下多囊肾分期。1、发生期:此...
- 2016-04-25
- 2016-04-25
- 2016-04-25
- 2016-04-25
- 2016-04-25
- 2016-04-25
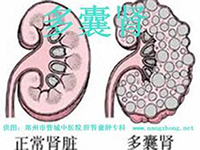





- 热门阅读



- 热点排行
- 多囊肾的早期症状有什么
- 得了多囊肾会有哪些症状
- 日常生活中多囊肾患者应注意什么?
- 多囊肾怎样治疗是有效果的
- 多囊肾的发病主要特点是什么
- 多囊肾注意什么你知道吗?
- 多囊肾的预防方法
- 多囊肾容易并发哪些疾病?
免费提问

